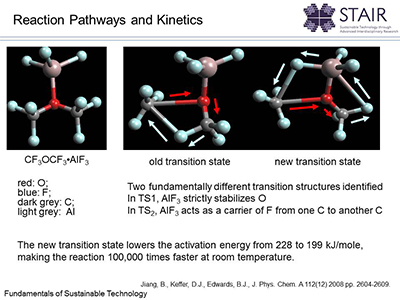
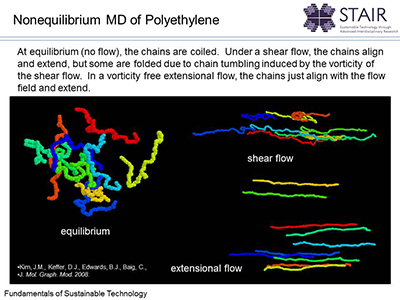
The Keffer research group at the University of Tennessee uses
multiscale modeling techniques to develop relationships between
the molecular structure of materials and their corresponding properties.
Research Outline
This research description is organized into sections on our broad approach to research,
specific materials under investigation and computational techniques employed.
- Research Approach
- Materials of Interest
- Research Methods
I. Research Approach
- Sustainable Energy
- Materials Discovery
- Experimental Collaboration
- Neutron Characterization
I.A. Sustainable Energy:
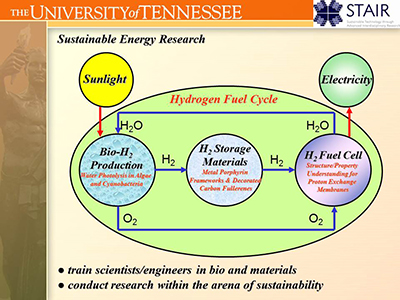 The defining challenge of our time is to find a way to bring the health and prosperity
of modern, developed countries to the entire global population in a manner that
allows future generations to enjoy the same environment and natural resources of the Earth that our current generation enjoys.
In the modern world, quality of life is highly correlated with energy usage.
Thus a critical component of this humanitarian challenge is technical in nature.
Furthermore, the transformational technologies that will allow for sustainable energy production
from renewable resources hinge on the materials underlying the technology, including solar panels, polymeric fuel cell membranes,
battery electrodes, biomass catalysts or nanostructured membranes for separation.
In the UT Computational Materials Research Group, we are contributing to the solution of this primary challenge of the twenty-first century
through the investigation of structure/property relationship in materials with sustainable energy applications.
The defining challenge of our time is to find a way to bring the health and prosperity
of modern, developed countries to the entire global population in a manner that
allows future generations to enjoy the same environment and natural resources of the Earth that our current generation enjoys.
In the modern world, quality of life is highly correlated with energy usage.
Thus a critical component of this humanitarian challenge is technical in nature.
Furthermore, the transformational technologies that will allow for sustainable energy production
from renewable resources hinge on the materials underlying the technology, including solar panels, polymeric fuel cell membranes,
battery electrodes, biomass catalysts or nanostructured membranes for separation.
In the UT Computational Materials Research Group, we are contributing to the solution of this primary challenge of the twenty-first century
through the investigation of structure/property relationship in materials with sustainable energy applications.
I.B. Materials Discovery:
Modern materials discovery is no longer a process of trial and error. Rather, an interdisciplinary team involving materials synthesizers,
specialists in materials characterization and materials modelers work together in an integrated manner.
The materials synthesizers make new materials, which are characterized both in terms of their structure and properties of interest.
The modelers provide a molecular-level understanding for the relationship between the observed structure and properties.
This understanding is communicated back to the synthesizers who target the synthesis of their next iteration of the material
in order to capture a specific structure, which will ideally, possess the desired properties for optimal performance.
Researchers in the UT Computational Materials Research Group perform modeling as part of this materials discovery cycle.
I.C. Experimental Collaboration:
 In the materials discovery process, it is essential that the modeler and experimentalist work in close collaboration for several reasons.
First, the experimentalist must be able to exercise precise control over the structure of interest in the synthesized materials.
If, for example, there is a broad spectrum of active sites, any model of a single site will likely fail to provide meaningful comparisons.
Second, if there is to be meaningful comparison between the computational and experimental results,
the experimentalist must participate in the model formulation.
A model that fails to correspond to the synthesized system is of limited utility in understanding the behavior of that material.
Third, the modeler must have access to experimental data for the properties of interest to validate the model for a test set of materials.
Finally, the modeler must communicate the modeling results to the experimentalist, who can make practical use of them in the synthesis of new materials.
The strategy of the UT Computational Materials Research Group adheres to each of these principles.
In the materials discovery process, it is essential that the modeler and experimentalist work in close collaboration for several reasons.
First, the experimentalist must be able to exercise precise control over the structure of interest in the synthesized materials.
If, for example, there is a broad spectrum of active sites, any model of a single site will likely fail to provide meaningful comparisons.
Second, if there is to be meaningful comparison between the computational and experimental results,
the experimentalist must participate in the model formulation.
A model that fails to correspond to the synthesized system is of limited utility in understanding the behavior of that material.
Third, the modeler must have access to experimental data for the properties of interest to validate the model for a test set of materials.
Finally, the modeler must communicate the modeling results to the experimentalist, who can make practical use of them in the synthesis of new materials.
The strategy of the UT Computational Materials Research Group adheres to each of these principles.
I.D. Neutron Characterization:
 Neutron scattering is a powerful tool to characterize atomic structure and dynamics with high precision.
It is a tool of choice especially for complex and hierarchical materials,
because modern neutron techniques allow the structure to be studied over various length-scales, from local, mesoscopic to the macroscale.
Neutron scattering is also advantageous because the properties of materials we study are often determined by light elements,
such as hydrogen and oxygen, which are difficult to study by x-rays.
In addition, because of the high penetration capability of neutrons it is a unique method for the study of materials in action,
in-situ and under operando conditions, to see how materials behave in the realistic environment.
The presence of the Spallation Neutron Source (SNS) in Oak Ridge, Tennessee allows modelers in the UT Computational Materials Research Group
to combine state-of-the-art computational techniques with the world's best neutron source in order to
characterize the structure and dynamics of new materials with unprecedented sophistication and accuracy.
Neutron scattering is a powerful tool to characterize atomic structure and dynamics with high precision.
It is a tool of choice especially for complex and hierarchical materials,
because modern neutron techniques allow the structure to be studied over various length-scales, from local, mesoscopic to the macroscale.
Neutron scattering is also advantageous because the properties of materials we study are often determined by light elements,
such as hydrogen and oxygen, which are difficult to study by x-rays.
In addition, because of the high penetration capability of neutrons it is a unique method for the study of materials in action,
in-situ and under operando conditions, to see how materials behave in the realistic environment.
The presence of the Spallation Neutron Source (SNS) in Oak Ridge, Tennessee allows modelers in the UT Computational Materials Research Group
to combine state-of-the-art computational techniques with the world's best neutron source in order to
characterize the structure and dynamics of new materials with unprecedented sophistication and accuracy.
II. Materials of Interest

In the Computational Materials Research Group, we investigate structure/property relationships across a range of materials.
Some of the materials investigated of recent and current interest are described below.
- Proton Exchange Membranes for Fuel Cells
- Battery Anodes
- Nanoporous Catalysts
- Fuel Cell Catalyst Layers
- Metal Organic Nanotubes
II.A. Proton Exchange Membranes for Fuel Cells:
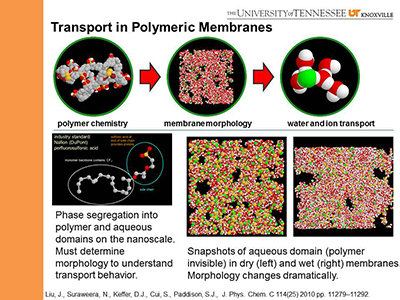 A primary expense of fuel cells is the noble metal catalyst.
Attempts to either reduce or replace the noble metal catalyst center on elevated temperatures during fuel cell operation.
However, the industry standard proton exchange membrane (PEM), Nafion,
a critical component of the fuel cell becomes dehydrated at higher temperatures, eventually becoming non-conductive.
The search for a high-temperature replacement for Nafion drives PEM research.
In collaboration with Prof. Jimmy Mays (UT Chemistry/ORNL),
we have investigated the performance of novel PEMs, including those composed of xs-PCHD/PEG block copolymers.
We relate the observed conductivity to molecular mechanisms of proton transport that are strong functions of the
morphology of the nanoscale aqueous domains within the hydrated membrane.
A primary expense of fuel cells is the noble metal catalyst.
Attempts to either reduce or replace the noble metal catalyst center on elevated temperatures during fuel cell operation.
However, the industry standard proton exchange membrane (PEM), Nafion,
a critical component of the fuel cell becomes dehydrated at higher temperatures, eventually becoming non-conductive.
The search for a high-temperature replacement for Nafion drives PEM research.
In collaboration with Prof. Jimmy Mays (UT Chemistry/ORNL),
we have investigated the performance of novel PEMs, including those composed of xs-PCHD/PEG block copolymers.
We relate the observed conductivity to molecular mechanisms of proton transport that are strong functions of the
morphology of the nanoscale aqueous domains within the hydrated membrane.
II.B. Lithium Ion Battery Anodes:
 Processing and materials are a primary expense in the manufacture of lithium ion batteries.
At Oak Ridge National Laboratory, Dr. Orlando Rios has led an effort to synthesize high-performance
battery anodes from lignin biomass through a very simplified process, relative to currently used materials.
The UT Computational Materials Research Group models these battery materials to understand the structure/property
relationships that lead to high ion capacity and high charging rates. This project also produces pair correlation functions
that can be directly compared with and thus explain spectra obtained from the NOMAD beamline at the Spallation Neutron Source.
Processing and materials are a primary expense in the manufacture of lithium ion batteries.
At Oak Ridge National Laboratory, Dr. Orlando Rios has led an effort to synthesize high-performance
battery anodes from lignin biomass through a very simplified process, relative to currently used materials.
The UT Computational Materials Research Group models these battery materials to understand the structure/property
relationships that lead to high ion capacity and high charging rates. This project also produces pair correlation functions
that can be directly compared with and thus explain spectra obtained from the NOMAD beamline at the Spallation Neutron Source.
II.C. Nanoporous Catalysts:
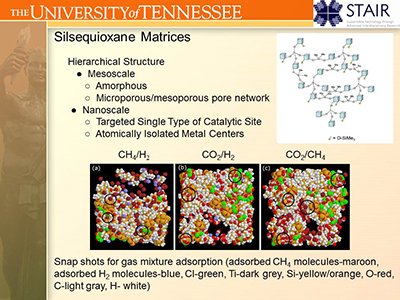 Catalysis is a field in which the ability to predict the catalytic performance of a material has lagged behind progress in other fields,
in part due to the difficulty of synthesizing catalysts with a single type of active site. Recent advances in synthetic procedures
by the group of Prof. Craig Barnes (UT Chemistry) have provided a path for a class of nanoporous adsorbents in which the nanoscale structure
of a single type of active site can be carefully controlled.
The UT Computational Materials Research Group models these materials in order to understand how the adsorptive, diffusive and catalytic
properties of materials within this class of catalysts are functions of the nanoscale and mesoscale structure.
Catalysis is a field in which the ability to predict the catalytic performance of a material has lagged behind progress in other fields,
in part due to the difficulty of synthesizing catalysts with a single type of active site. Recent advances in synthetic procedures
by the group of Prof. Craig Barnes (UT Chemistry) have provided a path for a class of nanoporous adsorbents in which the nanoscale structure
of a single type of active site can be carefully controlled.
The UT Computational Materials Research Group models these materials in order to understand how the adsorptive, diffusive and catalytic
properties of materials within this class of catalysts are functions of the nanoscale and mesoscale structure.
II.D. Fuel Cell Catalyst Layers:
 Fuel cell performance critically depends upon the peformance of catalyst nanoparticles.
In a fuel cell anode catalyst layer, the nanoparticles must dissociate molecular hydrogen into protons and electrons.
Thus each functioning nanoparticle must participate in three transport processes (in addition to its primary catalytic role).
First gaseous hydrogen must reach the nanoparticle. Second, protons must be passed through an ionomer film to the PEM.
Third, the electrons must be passed to the carbon support to the rest of the electrical circuit. A catalyst nanoparticle
that cannot participate in any one of these three transport processes is wasted. In this work, the UT Computational Materials Research Group
collaborated with microcopist Prof. David Joy (UT Materials Science & Engineering/ORNL) to investigate how the structure and composition
of the catalyst layer impacts the ability of catalyst nanoparticles to properly function.
Fuel cell performance critically depends upon the peformance of catalyst nanoparticles.
In a fuel cell anode catalyst layer, the nanoparticles must dissociate molecular hydrogen into protons and electrons.
Thus each functioning nanoparticle must participate in three transport processes (in addition to its primary catalytic role).
First gaseous hydrogen must reach the nanoparticle. Second, protons must be passed through an ionomer film to the PEM.
Third, the electrons must be passed to the carbon support to the rest of the electrical circuit. A catalyst nanoparticle
that cannot participate in any one of these three transport processes is wasted. In this work, the UT Computational Materials Research Group
collaborated with microcopist Prof. David Joy (UT Materials Science & Engineering/ORNL) to investigate how the structure and composition
of the catalyst layer impacts the ability of catalyst nanoparticles to properly function.
II.E. Metal Organic Nanotubes:
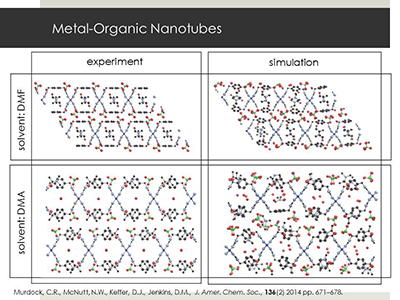 The nanoscale design of materials that targeted for a specific application is not yet widely realized.
However, nowhere is the concept more realizable than in Metal Organic Frameworks (MOFs), in which precise control over metal and organic components
has led to the prediction of hundreds of thousands of nanoporous crystalline materials.
Prof. David Jenkins (UT Chemistry) leads an effort to synthesize Metal Organic Nanotubes, which not only have the property that their
structure can be controlled through choice of components but also have the ability to change structure upon the introduction of some external stimulus.
This change in structure can take the form of molecular valves opening and closing. The UT Computational Materials Research Group
collaborates with Dr. Jenkins to expedite the process of materials discovery through the computational identification of best-choice components for
specific applications.
The nanoscale design of materials that targeted for a specific application is not yet widely realized.
However, nowhere is the concept more realizable than in Metal Organic Frameworks (MOFs), in which precise control over metal and organic components
has led to the prediction of hundreds of thousands of nanoporous crystalline materials.
Prof. David Jenkins (UT Chemistry) leads an effort to synthesize Metal Organic Nanotubes, which not only have the property that their
structure can be controlled through choice of components but also have the ability to change structure upon the introduction of some external stimulus.
This change in structure can take the form of molecular valves opening and closing. The UT Computational Materials Research Group
collaborates with Dr. Jenkins to expedite the process of materials discovery through the computational identification of best-choice components for
specific applications.
III. Research Methods
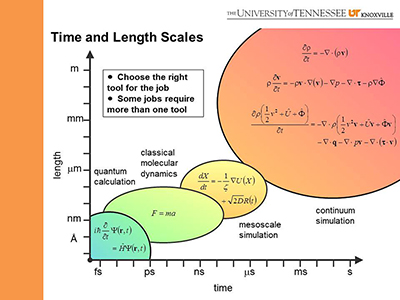
In the Computational Materials Research Group, we employ a suite of integrated tools
to describe phenomena in materials across four scales. Where established techniques are sufficient, we use them.
Where an application requires computational tools that do not yet exist, we develop and implement them.
- Quantum Scale
- Molecular Scale
- Mesoscale
- Macroscale
 University of Tennessee
University of Tennessee